ALLEGATO Xa"
Allegato Xa
Analisi gascromatografica degli esteri metilici degli acidi grassi1. OGGETTO
Il presente metodo d� un orientamento generale per l'applicazione della gascromatografia, con l'uso di colonne a riempimento o capillari, per determinare la composizione qualitativa e quantitativa di una miscela di esteri metilici degli acidi grassi ottenuta in conformit� con il metodo specificato nell'allegato Xb. Il metodo non � applicabile agli acidi grassi polimerizzati.
2. REAGENTI
- 2.1. Gas vettore
Gas inerte (azoto, elio, argo, idrogeno, ecc.) completamente essiccato ed avente un tenore di ossigeno inferiore a 10 mg/kg.
Nota 1: L'idrogeno, che viene usato come gas vettore soltanto con colonne capillari, pu� raddoppiare la velocit� dell'analisi, ma � pericoloso; sono disponibili dispositivi di sicurezza. - 2.2. Gas ausiliari
- 2.2.1. Idrogeno (purezza >99,9 %), esente da impurezze organiche.
- 2.2.2. Aria od ossigeno, esente da impurezze organiche.
- 2.3. Standard di riferimento
Miscela di esteri metilici di acidi grassi puri oppure esteri metilici di un grasso a composizione nota, di preferenza analogo a quello della sostanza grassa da analizzare. Sar� necessario l'ossidazione degli acidi grassi poliinsaturi.
Le istruzioni precisano che deve essere usata la consueta apparecchiatura per gascromatografia, facendo uso di colonne a riempimento e/o capillari nonch� di un rivelatore a ionizzazione di fiamma. � consigliabile usare apparecchiatura in grado di garantire l'efficienza e la risoluzione specificate al punto 4.1.2.
- 3.1. Gascromatografo
Il gascromatografo deve comprendere i seguenti elementi.- 3.1.1. Sistema ad iniezione
Usare un sistema ad iniezione:
a) con colonne a riempimento, aventi lo spazio morto minore possibile (in questo caso ilsistema di iniezione deve poter essere riscaldato a una temperatura di 20-50�C superiore a quella della colonna)
oppure
b) con colonne capillari, nel qual caso il sistema di iniezione deve essere appositamente progettato. Esso pu� essere del tipo a separazione oppure del tipo non a separazione sull'iniettore della colonna.
Nota 2: In assenza di acidi grassi aventi meno di 16 atomi di carbonio, pu� essere usatoun iniettore ad ago mobile. - 3.1.2. Stufa
La stufa deve essere atta a scaldare la colonna ad almeno 260�C e a mantenere la temperatura desiderata con l'approssimazione di 1�C per la colonna a riempimento e con l'approssimazione di 0,1�C per la colonna capillare. Quest'ultimo requisito � particolarmente importante se si usa una provetta di silice fusa. Il ricorso al riscaldamento programmato � raccomandato in tutti i casi e in particolare per gli acidi grassi aventi meno di 16 atomi di carbonio. - 3.1.3. Colonna e riempimento
- 3.1.3.1. Colonna costruita in materiale inerte alle sostanze da analizzare (cio� vetro o acciaio inossidabile) e avente le seguenti dimensioni:
a) lunghezza: 1-3 m. Se sono presenti acidi grassi a catena lunga (oltre C20) dovr� essere usata una colonna relativamente corta. Se invece si analizzano acidi a 4-6 atomi di carbonio, si raccomanda di usare una colonna avente una lunghezza di 2 m.
b) diametro interno: da 2 a 4 mm.
Nota 3: Se sono presenti componenti poliinsaturi aventi pi� di tre legami doppi, essi potranno essere decomposti in una colonna di acciaio inossidabile.
Nota 4: Pu� essere usato un sistema di colonne a riempimento gemelle.
- 3.1.3.2. Riempimento, comprendente i seguenti elementi:
a) supporto: terra di diatomee lavata con acido e silanizzata, oppure altro idoneo supporto inerte con una gamma ristretta di granulometria (compresa tra 125 e 200 �m, con variazioni di � 25 �m); la granulometria media � correlata al diametro interno e alla lunghezza della colonna;
b) fase stazionaria: tipo di poliestere di liquido polare (ad es. polisuccinato di dietilenglicole, polisuccinato di butandiolo, poliadipato di etilenglicole ecc.), cianosiliconi o qualsiasi altro liquido che consenta la separazione cromatografica richiesta (vedi clausola 5). La fase stazionaria deve essere compresa tra il 5 % (m/m) e il 20 % (m/m) del riempimento. Per alcune separazioni pu� essere usata una fase stazionaria non polare.
- 3.1.3.3. Condizionamento della colonna
Con la colonna staccata, dalla parte del rivelatore, scaldare gradualmente la stufa a 185�C e far passare una corrente di gas inerte attraverso la colonna di recente preparata ad un flusso compreso tra 20 ml/min e 60 ml/min per almeno 16 h a questa temperatura e per ulteriori 2 h a 195�C.
- 3.1.3.1. Colonna costruita in materiale inerte alle sostanze da analizzare (cio� vetro o acciaio inossidabile) e avente le seguenti dimensioni:
- 3.1.4. Colonna capillare
- 3.1.4.1. Tubo costituito di materiale inerte alle sostanze da analizzare (di solito vetro o silice fusa). Il diametro interno deve essere compreso tra 0,2 mm e 0,8 mm. La superficie interna deve esser sottoposta a un opportuno trattamento (ad es. preparazione della superficie, inattivazione) prima di essere ricoperto con la fase stazionaria. Nella maggior parte dei casi � sufficiente una lunghezza di 25 mm.
- 3.1.4.2. Fase stazionaria, di solito del tipo poliglicole [poli(etilenglicole) 20 000], poliestere (polisuccinato di butandiolo) oppure
polisilossano polare (cianosiliconi). Sono adatte le colonne legate (cross-linked).
Nota 5: Vi � il rischio che i polisilossani polari creino difficolt� nell'identificazione e separazione dell'acido linolenico e degli acidi a C20. Le coperture devono essere sottili, ad esempio 0,1 �m-0,2 �m. - 3.1.4.3. Montaggio e condizionamento della colonna
Osservare le normali precauzioni necessarie per il montaggio delle colonne capillari, ovvero sistemazione della colonna nella stufa (supporto), scelta e collegamento di giunti (a tenuta stagna), sistemazione delle estremit� della colonna nell'iniettore e nel rivelatore (riduzione degli spazi morti). Sottoporre la colonna ad un flusso di gas vettore [ad es. 0,3 bar (30 kPa) per una colonna avente una lunghezza di 25 mm e un diametro interno di 0,3 mm]. Condizionare la colonna programmando la temperatura della stufa a 3�C/min dalla temperatura ambiente a una temperatura di 10�C inferiore al limite di decomposizione della fase stazionaria. Mantenere la stufa a questa temperatura per 1 h fino a stabilizzazione della linea di base. Riportarla a 180�C in modo da lavorare in condizioni di isotermicit�.
Nota 6: Sono disponibili in commercio opportune colonne precondizionate.
- 3.1.5. Rivelatore, di preferenza idoneo ad essere riscaldato a una temperatura superiore a quella della colonna.
- 3.1.1. Sistema ad iniezione
- 3.2. Siringa
La siringa deve avere una capacit� massima di 10 �l ed essere graduata in divisioni di 0,1 �l. - 3.3. Registratore
Se la curva di registrazione deve essere usata per calcolare la composizione della miscela analizzata, � necessario un registratore elettronico di alta precisione compatibile con l'apparecchiatura usata. Detto registratore deve avere le seguenti caratteristiche:
a) tasso di risposta, al di sotto di 1,5 s, di preferenza 1 s (il tasso di risposta � il periodo necessario affinch� la punta di registrazione passi dallo 0 % al 90 % a seguito dell'introduzione improvvisa di un segnale del 100 %);
b) ampiezza della carta, minimo 20 cm;
c) velocit� della carta, adattabile a valori compresi tra 0,4 cm/min e 2,5 cm/min. - 3.4. Integratore o calcolatore (facoltativo)
Un calcolo rapido ed accurato pu� essere effettuato con l'ausilio di un integratore o calcolatore elettronico. Quest'ultimo deve dare una risposta lineare con una sensibilit� adeguata e la correzione della deviazione della linea di base deve essere soddisfacente.
Nelle operazioni descritte dal paragrafo 4.1 al paragrafo 4.3 si fa accenno all'uso di un rivelatore a ionizzazione di fiamma. Come alternativa pu� essere usato un gascromatografo munito di catarometro (che funziona in base al principio della variazione di conducibilit� termica). Le condizioni di funzionamento vengono pertanto modificate come descritto nella clausola 6.
- 4.1. Condizioni dell'analisi
- 4.1.1. Selezione delle condizioni operative ottimali
- 4.1.1.1. Colonna a riempimento
Nella scelta delle condizioni per effettuare la prova, devono essere prese in considerazione le seguenti varianti:
a) la lunghezza ed il diametro della colonna;
b) la natura e la quantit� della fase stazionaria;
c) la temperatura della colonna;
d) il flusso di gas vettore;
e) la risoluzione necessaria;
f) le dimensioni del campione da analizzare, scelto in modo che il collegamento tra il rivelatore e l'elettrometro diano una risposta lineare;
g) la durata dell'analisi.
In linea di massima i valori riportati nella tabella 1 e nella tabella 2 danno i risultati auspicati, cio� almeno 2 000 piatti teorici per metro di lunghezza della colonna per quanto si riferisce allo stearato di metile, e l'eluizione dello stesso entro 15 minuti circa. Se l'apparecchiatura lo consente, l'iniettore deve trovarsi a una temperatura di circa 200�C ed il rivelatore a una temperatura pari o superiore a quella della colonna. Di norma, il rapporto tra il flusso dell'idrogeno fornito al rilevatore a ionizzatore di fiamma e quello del gas vettore varia tra 1:2 e 1:1 in funzione del diametro della colonna. Il flusso dell'ossigeno � di circa 5-10 volte quello dell'idrogeno.
Tabella 1 Diametro interno della colonna mm Flusso del gas vettore ml/min 2 da 15 a 25 3 da 20 a 40 4 da 40 a 60
Tabella 2 Concentrazione della fase stazionaria % (m/m) Temperatura della colonna �C 5 175 10 180 15 185 20 185 - 4.1.1.2. Colonna capillare
Le caratteristiche di efficienza e di permeabilit� delle colonne capillari indicano che la separazione tra i costituenti e la durata dell'analisi sono ampiamente dipendenti dal flusso del gas vettore nella colonna. � pertanto necessario ottimizzare le condizioni operative influenzando questo parametro (o pi� semplicemente la pressione di testa della colonna), a seconda che si desideri migliorare le separazioni o effettuare un'analisi rapida.
- 4.1.1.1. Colonna a riempimento
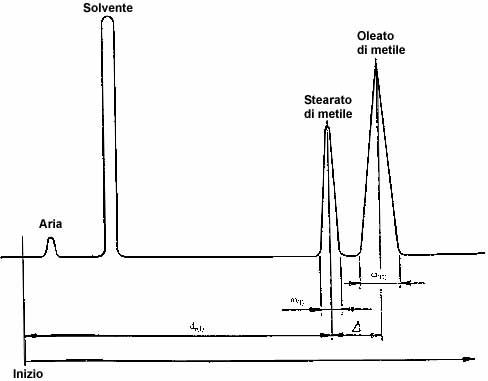
- 4.1.2. Determinazione del numero di piatti teorici (efficacia) e risoluzione (vedi figura 1)
Effettuare l'analisi di una miscela di stearato di metile e di oleato di metile in proporzioni pi� o meno equivalenti (ad es. esteri metilici del burro di cacao). Scegliere la temperatura della colonna e il flusso di gas vettore in modo che il massimo del picco dello stearato di metile venga registrato circa 15 minuti dopo il picco del solvente. Usare un quantitativo sufficiente della miscela di esteri metilici in modo che il picco dello stearato di metile occupi circa tre quarti della scala intera. Calcolare il numero dei piatti teorici, n (efficienza), con la formula:n = 16 {[dr (I) H] / [W (I)]}�
e la risoluzione R usando la formula:R = 2D / [W(I) + W (II)]
dove:
dr (I) = � la distanza di ritenzione, in millimetri, dall'inizio del cromatogramma fino al massimo del picco dello stearato di metile;
W(I) e W(II) = sono le ampiezze, in millimetri, del picchi rispettivamente dello stearato di metile e dell'oleato di metile, misurati tra i punti d'intersezione delle tangenti ai punti di inflessione della curva con la linea di base;
D = � la distanza, in millimetri, tra i picchi massimi dello stearato di metile e dell'oleato di metile.
Figura 1
Cromatogramma per la determinazione del numero di piatti teorici
(efficienza) e risoluzione
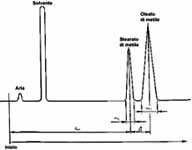
(un click sull'immagine per ingrandirla)
Le condizioni operative da scegliere sono quelle idonee ad almeno 2 000 piatti teorici per metro di lunghezza della colonna per quanto si riferisce allo stearato di metile e una risoluzione di almeno 1,25.
- 4.1.1. Selezione delle condizioni operative ottimali
- 4.2. Sostanza da analizzare
Usando la siringa (3.2) prendere 0,1 �l-2 �l della soluzione di esteri metilici preparata conformemente all'allegato Xb e iniettarli nella colonna. Nel caso di esteri non in soluzione, preparare una soluzione di circa 100 mg/ml in eptano di qualit� cromatografica ed iniettare da 0,1 �l a 1 �l di questa soluzione. Se l'analisi riguarda costituenti presenti soltanto in tracce, la quantit� di sostanza da analizzare pu� essere aumentata (fino a 10 volte).
- 4.3. Analisi
Di norma le condizioni operative sono quelle di cui al paragrafo 4.1.1. � possibile tuttavia operare a una temperatura inferiore della colonna quando si tratta di determinare acidi grassi aventi meno di 12 atomi di carbonio oppure a una temperatura pi� elevata quando si tratta di determinare acidi grassi con oltre 20 atomi di carbonio. All'occasione, � possibile programmare la temperatura in entrambi questi casi. Ad esempio, se il campione contiene gli esteri metilici degli acidi grassi con meno di 12 atomi di carbonio, iniettare il campione a 100�C (oppure da 50�C a 60�C se � presente acido butirrico) e raggiungere immediatamente la temperatura a un tasso compreso tra 4�C/min e 8�C/min in condizioni ottimali. In taluni casi possono essere associati i due procedimenti. Dopo il riscaldamento programmato, continuare l'eluizione a temperatura costante finch� tutti i componenti sono stati eluiti. Se lo strumento non prevede il riscaldamento programmato, usarlo a due temperature fisse comprese tra 100�C e 195�C. Se necessario, si raccomanda di effettuare un'analisi su due fasi fisse a polarit� differente per verificare l'assenza di picchi mascherati, ad esempio nel caso della presenza contemporanea di C18:3 e C20:0 oppure C18:3 e C18:2 associati. - 4.4. Preparazione del cromatogramma di riferimento e dei grafici di riferimento.
Analizzare la miscela standard di riferimento (2.3) nelle stesse condizioni operative di quelle usate per il campione e misurare i tempi di ritenzione o le distanze di ritenzione per gli acidi grassi costituenti. Su carta semilogaritmica, per qualsiasi grado di insaturazione, costruire i grafici che mostrano il logaritmo del tempo o della distanza di ritenzione in funzione del numero di atomi di carbonio. In condizioni isotermiche, i grafici relativi ad acidi a catena lineare aventi lo stesso grado di insaturazione devono essere linee rette. Queste linee devono essere all'incirca parallele. � necessario evitare condizioni in cui si possano verificare i "picchi mascherati", ovvero nei quali la risoluzione � insufficiente a separare due costituenti.
- 5.1. Analisi qualitativa
Identificare i picchi dell'estere metilico del campione dai grafici preparati al paragrafo 4.4, se necessario per interpolazione.
- 5.2. Analisi quantitativa
- 5.2.1. Determinazione della composizione
A parte casi eccezionali, usare il metodo di normalizzazione interno, cio� presumere che la totalit� dei componenti del campione siano rappresentati sul cromatogramma, in modo che il totale delle aree sotto i picchi costituisca il 100 % dei costituenti (eluizione totale). Se nell'apparecchiatura � previsto un integratore, usare i dati da esso ottenuti. In caso negativo, determinare l'area sotto ciascun picco moltiplicando l'altezza del picco per l'ampiezza a met� altezza e, se necessario, prendere in considerazione le varie attenuazioni usate durante la registrazione. - 5.2.2. Metodo di calcolo
- 5.2.2.1. Caso generale
Calcolare il contenuto di un dato componente I, espresso come percentuale in massa degli esteri metilici, determinando la percentuale rappresentata dall'area del picco corrispondente relativa alla somma delle aree di tutti i picchi, usando la formula seguente:(� A / Ai) * 100
dove:
Ai = � l'area sotto il picco corrispondente al componente i;
� A = � la somma delle aree sotto tutti i picchi.
Esprimere il risultato con l'approssimazione di una decimale.
Nota 7: In questo caso generale, si ritiene che il risultato del calcolo basato sulle aree relative rappresenti una percentuale in massa. Per i casi nei quali questa asserzione non � giustificata, vedi 5.2.2.2. - 5.2.2.2. Uso dei fattori di correzione
In taluni casi, ad esempio in presenza di acidi grassi aventi meno di 8 atomi di carbonio oppure di acidi aventi gruppi secondari, quando si usano rivelatori di conduttivit� termica oppure quando � necessario il grado pi� elevato di accuratezza, devono essere usati fattori di correzione che convertano le percentuali delle aree e dei picchi in percentuali in peso dei componenti. Determinare i fattori di correzione con l'ausilio di un cromatogramma derivato dall'analisi di una miscela di riferimento di esteri metilici di composizione nota, effettuata in condizioni operative identiche a quelle usate per il campione. Per questa miscela di riferimento, la percentuale in peso del componente i � data dalla formula:(mi / � m) * 100
dove:
mi = � il peso del componente i nella miscela di riferimento;
� m = � il totale delle masse dei vari componenti della miscela di riferimento.
Dal cromatogramma della miscela di riferimento (4.4) calcolare la percentuale (area/area) del componente i come segue:(Ai / � A) * 100
dove:
Ai � l'area sotto il picco corrispondente al componente i,
� A � la somma delle aree sotto tutti i picchi.
Il fattore di correzione viene quindi calcolato come segue:Ki = (mi * � A) / (Ai * � m)
Di norma i fattori di correzione vengono espressi facendo riferimento a KC16 sicch� i fattori relativi diventano:K'i = (Ki / KC 1 6)
Per il campione il contenuto di ciascun componente espresso come percentuale in degli esteri metilici �:(K'i * Ai) / [� (K' i * Ai)] * 100
Esprimere i risultati con l'approssimazione di una decimale. - 5.2.2.3. Uso di uno standard interno
In alcune analisi (ad es. quando non tutti gli acidi grassi sono quantificati, ad esempio quando sono presenti acidi con 4 e 6 atomi di carbonio accanto ad acidi con 16 e 18 atomi di carbonio, oppure quando � necessario determinare il quantitativo assoluto di un acido grasso in un campione) � necessario ricorrere ad uno standard interno. Vengono spesso usati acidi grassi con 5,15 o 17 atomi di carbonio. Deve essere determinato l'eventuale fattore di correzione per lo standard interno. La percentuale in peso del componente i, espressa come esteri metilici, � data dalla formula:(ms * K'i * Ai) / (m * K's * As) * 100
dove:
Ai � l'area sotto il picco corrispondente al componente i;
As � l'area sotto il picco corrispondente allo standard interno;
K'i � il fattore di correzione per il componente i (relativo a KC 1 6);
K's � il fattore di correzione dello standard interno (relativo a KC 1 6);
m � il peso, in milligrammi, della sostanza da analizzare;
ms � il peso, in milligrammi, dello standard interno.
Esprimere i risultati con l'approssimazione di una decimale.
- 5.2.2.1. Caso generale
- 5.2.1. Determinazione della composizione
Per la determinazione della composizione qualitativa e quantitativa di una miscela di esteri metilici degli acidi grassi pu� essere usato altres� un gascromatografo associato a un rivelatore funzionante in base al principio dei mutamenti di conduttivit� termica (catarometro). Se quest'ultimo viene usato, le condizioni specificate alle clausole 3 e 4 devono essere modificate come indicato nella tabella 3. Per l'analisi quantitativa, usare i fattori di correzione definiti al paragrafo 5.2.2.2.
| Variabile | Valore/condizioni |
| Colonna | Lunghezza: da 2 m a 4 m Diametro interno: 4 mm |
| Supporto | Granulometria compresa tra 160 �m e 200 �m |
| Concentrazione della fase stazionaria | Dal 15 % (m/m) al 25 % (m/m) |
| Gas vettore | Elio oppure, in mancanza di quest'ultimo, idrogeno, col tenore pi� basso possibile di ossigeno |
| Gas ausiliari | Nessuno |
| Temperatura dell'iniettore | Da 40�C a 60�C al di sopra di quella della colonna |
| Temperatura della colonna | Da 180�C a 200�C |
| Flusso del gas vettore | Di norma tra 60 ed 80 ml/min |
| Entit� del campione di sostanza iniettato | Di norma tra 0,5 �l e 2�l |
7. RELAZIONE SULLA PROVA
La relazione sulla prova deve specificare i metodi usati per la preparazione degli esteri metilici e per l'analisi gascromatografica nonch� i risultati tenuti. Essa deve citare altres� tutti i dettagli operativi non specificati nella presente norma internazionale oppure considerati come facoltativi, nonch� i particolari di qualsiasi evento che possa avere influenzato i risultati. La relazione sulla prova deve comprendere tutti i dati necessari per la completa identificazione del campione.

![]()